
A triagem para aminoacidopatias teve significativa melhora com a tecnologia da espectrometria de massas em tandem (MS/MS), especialmente para fenilcetonúria (PKU), doença da urina de xarope de bordo (MSUD) e homocistinúria. A análise dos aminoácidos neutros (com exceção da glicina) e ácidos pode ser feita concomitante à das acilcarnitinas, utilizando um tipo especial de triagem. O perfil dos aminoácidos neutros e ácidos, obtido de uma amostra de sangue em papel-filtro de um recém-nascido normal pode ser visto na fig. 4.
Chace et al. compararam a espectrometria de massas em tandem com o método fluorimétrico na triagem de PKU, e encontraram uma taxa de falso-positivos 100 vezes menor pela MS/ MS, devido à maior exatidão e precisão na mensuração da fenilalanina e à possibilidade de confirmação pela determinação da razão molar fenilalanina/tirosina. Os autores também constataram a utilidade do método na triagem neonatal de PKU em amostras coletadas precocemente (antes de 24 horas ou a partir de 8 horas). O nível de corte utilizado para fenilalanina na MS/MS é mais baixo que no método fluorimétrico, o que pode ser atribuído à presença de interferentes neste último, o que eleva a concentração de fenilalanina.
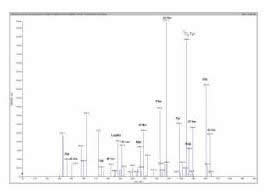
Fig. 4 Perfil de aminoácidos neutros, com exceção de glicina, e ácidos, após butilação ácida de extrato metanólico de amostra de sangue em papel-filtro de recém-nascido normal. Ala: alanina; Val: valina; Leu/Ile: leucina/ isoleucina; Met: metionina; Phe: fenilalanina; Tyr: tirosina; Asp: ácido aspártico; Glu: ácido glutâmico.
A MS/ MS também pode ser aplicada na triagem da MSUD através da mensuração de dois analitos (leucina + isoleucina e valina) e duas razões molares (leucina + isoleucina/ fenilalanina e valina/ fenilalanina). Deve-se aqui ser mencionada uma limitação da MS/ MS, que é a incapacidade de diferenciar compostos de mesma massa, o que é particularmente importante para a leucina, que tem três formas isoméricas principais (leucina, isoleucina e aloisoleucina) e também é isobárica com a hidroxiprolina. Isto não parece diminuir a utilidade da MS/ MS na triagem da MSUD, muito embora o potencial diagnóstico da aloisoleucina seja bastante expressivo.
A triagem de homocistinúria e de outras hipermetioninemias também pode ser feita por MS/ MS, através da avaliação da metionina e da razão molar metionina/ leucina + isoleucina.
O ciclo da uréia consiste em uma série de reações enzimáticas que convertem a amônia, liberada durante o catabolismo das proteínas, em uréia. A uréia, a principal escória nitrogenada, é então excretada na urina.
| Há cinco enzimas envolvidas no ciclo da uréia: carbamil-fosfato sintetase (CPS), ornitina-transcarbamilase (OTC), arginino-succinato sintetase (AS), arginino-succinato liase (AL), e arginase. |
Todos os distúrbios do ciclo da uréia resultam na hiperamonemia. Os níveis elevados de amônia plasmática são altamente neurotóxicos aos seres humanos.
Os pacientes com deficiência de AS têm níveis acentuadamente elevados de citrulina plasmática, enquanto os pacientes com deficiência de AL têm níveis moderadamente elevados de citrulina e um aumento de ácido arginino-succínico plasmático.
Os pacientes com deficiência de CPS e OTC possuem níveis baixos ou indetectáveis de citrulina plasmática, mas na deficiência de OTC ocorre um aumento do ácido orótico urinário. O ácido orótico resulta do transbordamento do excesso de carbamil-fosfato do ciclo da uréia para a via das pirimidinas.
Estima-se que os distúrbios do ciclo da uréia ocorram em 1 caso em 30.000 nascidos vivos, todos são herdados como traços autossômicos recessivos à exceção da deficiência da ornitina-transcarbamilase (OTC), que é herdada como um traço ligado ao X.
As famílias dos pacientes com distúrbios do ciclo da uréia devem receber aconselhamento genético, porque a detecção do portador e o diagnóstico pré-natal estão disponíveis para a maioria dos distúrbios.
Quase sempre o quadro clínico se apresenta durante o período neonatal por uma deterioração neurológica rapidamente progressiva que começa após um período de 1-2 dias de normalidade aparente. À medida que os níveis de amônia aumentam, os pacientes afetados desenvolvem recusa alimentar, anorexia, alterações do comportamento, irritabilidade, vômitos, letargia, ataxia, convulsões, coma, edema cerebral, e finalmente colapso circulatório. As formas menos severas podem apresentar-se em qualquer idade, até mesmo na fase adulta, e os pacientes apresentam sintomas intermitentes de hiperamonemia, transtornos do comportamento, ou disfunção neurológica.
Quando da apresentação inicial, devem ser coletadas amostras para perfil de aminoácidos quantitativo do plasma, análise de ácidos orgânicos urinários e dosagem do ácido orótico na urina. O padrão de alterações aponta geralmente a um distúrbio específico. A confirmação da deficiência enzimática suspeitada para muitos dos distúrbios, pode ser feita com eritrócitos periféricos ou com fibroblastos cultivados da pele; outros distúrbios requerem a biópsia hepática. Alguns dos distúrbios podem ser confirmados por estudos genéticos moleculares.
O principal diagnóstico diferencial dos distúrbios do ciclo da uréia num neonato com hiperamonemia são: a hiperamonemia transitória do recém-nascido e as acidemias orgânicas. A hiperamonemia transitória do recém-nascido, acomete bebês prematuros nas primeiras 24h de vida, enquanto que os neonatos com acidemias orgânicas classicamente se apresentam com acidose metabólica, “anion gap” elevado e cetonúria.
Aspectos Clínicos de Algumas das mais Importantes Aminoacidopatias e Distúrbios do Ciclo da Uréia
- Hiperfenilalaninemias
A elevação dos níveis de fenilalanina na triagem neonatal (Phe > 4,0 mg%) requer o diagnóstico diferencial entre as seguintes patologias:
| Fenilcetonúria clássica (PKU) | Deficiência grave da enzima fenilalanina-hidroxilase levando ao acúmulo de fenilalanina no sangue com graves sequelas neurológicas caso o tratamento não seja instituído precocemente. Os níveis de fenilalanina são muito elevados (> 10 mg%) e os de tirosina muito baixos. |
| Hiperfenilalaninemia Benigna | Deficiência menos acentuada da enzima fenilalanina-hidroxilase com elevação moderada dos níveis de fenilalanina (4,0 a 10mg%). Não há necessidade de tratamento dietoterápico. |
| Deficiência da Tetrahidrobiopterina (BH4) | BH4 é um cofator para as enzimas fenilalanina-hidroxilase, tirosina-hidroxilase e triptofano-hidroxilase. A deficiência deste cofator leva à deficiência na produção de neurotransmissores (DOPA e serotonina) com conseqüente lesão neurológica grave independente do tratamento dietoterápico. Os níveis de fenilalanina podem ser tão elevados quanto na fenilcetonúria clássica. |
| Tirosinemia | Ocorre elevação tanto da fenilalanina quanto da tirosina. A forma mais comum é a Tirosinemia transitória do recém-nascido que está freqüentemente relacionada à introdução precoce de leite de vaca, sendo também comum em prematuros e gemelares. Apesar de rara, deve-se incluir no diagnóstico diferencial a Tirosinemia tipo I que é uma doença grave que se manifesta geralmente nas primeiras semanas de vida com um quadro progressivo de insuficiência hepática, diátese hemorrágica, lesão renal (Síndrome de Fanconi), podendo levar ao óbito os casos não tratados. |
| Hiperfenilalaninemia secundária a lesão hepática de diversas causas. | Ocorre elevação da fenilalanina e da tirosina. |
- A fenilalanina pode ser dosada pelos métodos convencionais (método enzimático) mas a utilização da espectrometria de massas apresenta as seguintes vantagens:
| Diminuição de 50% nos índices de falso-positivos. |
| Através da análise das concentrações da fenilalanina e da tirosina e da relação fenilalanina/tirosina é possível fazer o diagnóstico diferencial entre as diversas causas de hiperfenilalaninemia. Apenas os pacientes com fenilcetonúria clássica e deficiência de BH4 apresentam uma relação Phe/Tir aumentada.o que orienta a indicação terapêutica. Os pacientes com Tirosinemia apresentam níveis muito elevados de tirosina e moderadamente elevados de fenilalanina. O diagnóstico de Tirosinemia tipo I requer a dosagem da succinilacetona em sangue ou urina. |
| Diagnóstico destas patologias pode ser feito precocemente (antes de 24 horas de vida) considerando a grande sensibilidade do método. |
- Doença da urina de xarope de bordo (MSUD)
MSUD é causada por metabolismo anormal de três aminoácidos de cadeia ramificada. A incidência é de 1 em 200.000 nascidos vivos. Os sintomas incluem um odor de xarope de bordo na urina, recusa alimentar, letargia, coma e retardo mental. A morte comumente ocorre nos três primeiros meses de vida. O tratamento requer restrição dietética de aminoácidos de cadeia ramificada, necessitando de uma fórmula complexa e acompanhamento médico rigoroso da criança.
- Hiperglicinemia não-cetótica (NKH)
É uma doença metabólica autossômica recessiva que se apresenta no período neonatal como um quadro neurológico grave, quase sempre fatal. Os sobreviventes apresentam déficit neurológico grave, retardo psicomotor e convulsões de difícil controle. A lesão metabólica da NKH deve-se à incapacidade do sistema de clivagem da glicina (GCS), em proceder a descarboxilação oxidativa da glicina, levando a um acúmulo deste aminoácido neurotransmissor, que resulta numa estimulação excessiva dos receptores N-metil-D-aspartato (NMDA) cerebrais. O tratamento inclui redução da sobrecarga de glicina e o uso de antagonistas dos efeitos neurotransmissores da glicina (antagonistas NMDA).
- Hipermetioninemia
Níveis sangüíneos elevados de metionina são encontrados na hipermetioninemia, na homocistinúria, e na tirosinemia tipo I. Na hipermetioninemia não é observado o aumento de tirosina encontrada na tirosinemia tipo I e nem o aumento de homocisteína observado na homocistinúria. A maior parte dos casos de hipermetioninemia é devida a uma deficiência de metionina-adenosiltransferase (MAT) I/III causada por mutações no gene MAT1A. Os efeitos clínicos e metabólicos destas mutações são muito variáveis. A maioria dos indivíduos são heterozigotos para uma mutação dominante, apresentando níveis plasmáticos moderadamente elevados de metionina e um quadro clínico benigno e assintomático. No entanto um pequeno grupo de indivíduos são homozigotos para mutações recessivas, apresentando uma redução significativa da atividade da MAT, níveis muito elevados de metionina plasmática e sintomas neurológicos, incluindo alterações da substância cinzenta e desmielinização cerebral.
- Homocistinúria
A causa mais comum de homocistinúria é uma deficiência da enzima cistationina sintetase. A incidência é de 1em 200.000 nascidos vivos. Entre os sintomas se encontram tromboembolismo, deslocamento do cristalino (que pode ocorrer mesmo com tratamento regular), escoliose, osteoporose, retardo mental, convulsões e distúrbios psiquiátricos. Aproximadamente 50 por cento dos indivíduos não tratados morrem antes de 25 anos de idade. O tratamento pode incluir uma dieta restrita de metionina, suplementada de cistina, assim como altas doses de Vitamina B6.
- Tirosinemia
É uma doença metabólica que causa elevação dos níveis sangüíneos de tirosina, um aminoácido presente na maioria das proteínas. A incidência é 1:100.000 nascidos vivos, com ambos os sexos igualmente afetados. Os sintomas incluem déficit ponderal, vômitos, diarréia, e um odor como de repolho. Outros sintomas incluem fígado aumentado, edema e risco de doença hemorrágica. Se não tratada, a tirosinemia é fatal dentro do primeiro ano de vida. O tratamento inclui restrições dietéticas.
- Acidúria arginino-succínica
Esta doença é extremamente rara. Os sintomas são hiperamonemia acompanhada por falta de apetite, vômitos, apatia, convulsões e coma. O início dos sintomas se dá normalmente no nascimento, mas podem não ser notados durante dias ou semanas. Sem tratamento, ocorrerá lesão cerebral, coma e morte. O tratamento inclui uma dieta de alto teor calórico, restrita em proteínas, suplementação de arginina e administração de benzoato de sódio e fenilacetato de sódio. A diálise pode ser necessária.
- Argininemia
Duas formas de arginase, A-I e A-II, são especificadas por genes distintos, ARG1 E ARG2. A isoenzima A-I contribui com 98% da atividade de arginase hepática e está ausente na argininemia. A doença se caracteriza por graus variados de hiperamonemia, paraplegia espástica com início aos 2 – 3 anos de idade, convulsões epilépticas e retardo mental de início mais tardio. Os níveis de arginina estão elevados no sangue e no líquido cefalorraquiano dos pacientes e intermediários nos heterozigotos. A atividade de arginase é muito baixa nos eritrócitos dos pacientes e intermediária nos heterozigotos. Pode haver um efeito colateral com o tratamento com valproato de sódio, um anticonvulsivante. A doença pode permanecer sem diagnóstico em casos de paralisia cerebral, devido aos seus sintomas relativamente mais leves do que nos outros distúrbios do ciclo da uréia, nos quais a hiperamonemia é muito mais severa. O tratamento é feito pela administração de benzoato e restrição de arginina.
- Citrulinemia
A citrulinemia origina-se de uma deficiência de arginino-succinato sintetase. A epidemiologia, os sintomas, e o tratamento são os mesmos da acidúria arginino-succínica.
- Síndrome de hiperornitinemia-hiperamonemia-homocitrulinúria
Os sintomas clínicos estão relacionados à hiperamonemia e se assemelham àqueles dos outros distúrbios do ciclo da uréia. A fisiopatologia da doença envolve uma diminuição do transporte de ornitina para as mitocôndrias, resultando num acúmulo de ornitina citoplasmática e numa redução da capacidade de processar sobrecargas de amônia. A ornitina-delta-aminotransferase, está normal. Não ocorrem problemas visuais ou alterações do fundo de olho como na ornitinemia com atrofia circular corio-retínica. Numa dieta restrita de proteína as concentrações plasmáticas de ornitina são geralmente mais baixas que na atrofia circular. Postula-se que a homocitrulina origine-se da transcarbamoilação da lisina. A herança é autossômica recessiva.